
Comparative analysis of prophages in Streptococcus mutans genomes
- Published
- Accepted
- Received
- Academic Editor
- Ramy Aziz
- Subject Areas
- Bioinformatics, Microbiology
- Keywords
- Comparative genomics, Streptococcus mutans, Prophages
- Copyright
- © 2017 Fu et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2017. Comparative analysis of prophages in Streptococcus mutans genomes. PeerJ 5:e4057 https://doi.org/10.7717/peerj.4057
Abstract
Prophages have been considered genetic units that have an intimate association with novel phenotypic properties of bacterial hosts, such as pathogenicity and genomic variation. Little is known about the genetic information of prophages in the genome of Streptococcus mutans, a major pathogen of human dental caries. In this study, we identified 35 prophage-like elements in S. mutans genomes and performed a comparative genomic analysis. Comparative genomic and phylogenetic analyses of prophage sequences revealed that the prophages could be classified into three main large clusters: Cluster A, Cluster B, and Cluster C. The S. mutans prophages in each cluster were compared. The genomic sequences of phismuN66-1, phismuNLML9-1, and phismu24-1 all shared similarities with the previously reported S. mutans phages M102, M102AD, and ϕAPCM01. The genomes were organized into seven major gene clusters according to the putative functions of the predicted open reading frames: packaging and structural modules, integrase, host lysis modules, DNA replication/recombination modules, transcriptional regulatory modules, other protein modules, and hypothetical protein modules. Moreover, an integrase gene was only identified in phismuNLML9-1 prophages.
Introduction
A prophage is a temperate bacteriophage genome integrated into a host bacterial DNA chromosome, which has the ability to enter a lysogenic state and replicate vertically with the host (St-Pierre & Endy, 2008). Prophages are an important source of virulence factors and other determinants that affect bacterial pathogenesis. Whole genome sequencing projects and comparative genomic analysis have revealed that prophage sequences are widespread among bacterial genomes, such as Moraxella catarrhalis (Ariff et al., 2015), Enterococcus spp. (Duerkop, Palmer & Horsburgh, 2014), Lactococcus spp. (Ventura et al., 2007), Mycobacterium spp. (Fan et al., 2014), and Streptococcus suis (Tang et al., 2013). Yet very little is known about Streptococcus mutans prophages.
Dental caries are the most prevalent dental disease and an important public health problem worldwide (Kidd & Fejerskov, 2013). The development of carious lesions stems from a dynamic process mediated by acid produced by cariogenic bacteria, such as Streptococcus mutans, Streptococcus sobrinus, and Lactobacilli, eventually resulting in de-mineralization and damage to the tooth structure (Argimon et al., 2014; Ericson et al., 2003). S. mutans are Gram-positive and biofilm-forming bacteria that can adhere to the tooth surface and contribute to dental plaque. S. mutans is the major pathogen responsible for dental caries in humans (Freires et al., 2017; Motegi et al., 2006). To the best of our knowledge, there have not been any reports describing S. mutans prophages, and only five S. mutans phages have been isolated. Three of them, M102, M102AD, and ϕAPCM01, have been sequenced (Dalmasso et al., 2015; Delisle et al., 2012; Van der Ploeg, 2007). Two other S. mutans phages, f1 and e10, have previously been isolated and tested for their host range and morphology, but not sequenced (Delisle & Rostkowski, 1993). Currently, there are 171 S. mutans genomic sequences in the National Center for Biotechnology Information (NCBI) database. Genomic sequencing of S. mutans has made it possible to identify prophages and perform comparative genomic analysis of prophage sequences and organization.
In this study, we screened all available complete S. mutans genomic sequences and identified 35 prophage-like elements present in these sequences. We also report the functional features of the intact prophages in comparison with another S. mutans phage, M102AD. Comparative genomic analysis and genome content analysis of S. mutans prophages were performed, and genetic information was analyzed.
Materials and Methods
Data collection and prophage sequence analyses
In total, 171 S. mutans genomes were obtained from NCBI. For prophage identification, tools such as PhiSpy (Akhter, Aziz & Edwards, 2012) and VirSorter (Roux et al., 2015) have been published as fast, relatively straight forward, and easier to use. We detected putative prophage DNA sequence data using the previously reported PHAge Search Tool Enhanced Release (PHASTER) method. PHASTER (http://phaster.ca/) was used to analyze bacterial genomes to identify and annotate putative prophage sequences (Arndt et al., 2016; Fan et al., 2016).
Genomic and comparative genomic analyses of S. mutans prophages
Dot plot comparisons of S. mutans prophage genomes were performed using Geneious v.10.0.5 (http://www.geneious.com, Kearse et al., 2012). Prophage open reading frames (ORFs) were predicted using PHASTER, GeneMarkS, and BLASP (Zhu, Lomsadze & Borodovsky, 2010). The genomic organization of S. mutans prophage genome maps was constructed by SnapGene or SnapGene Viewer (http://www.snapgene.com; GSL Biotech, Chicago, IL, USA) (Sturmberger et al., 2016). The genomes comparison was performed on the DNA level with BLASTn (http://blast.ncbi.nlm.nih.gov/Blast.cgi) based on the percentage of sequence identity, and results were analyzed using Artemis Comparison Tool software (Carver et al., 2008). Default settings were used in all software.
Phylogenetic analysis
Alignments of S. mutans phage and prophage genomic sequences were performed using MEGA version 7.0 (Tamura et al., 2007). Phylogenetic analysis was performed by the neighbor-joining (NJ) method and visualized using MEGA software. Phylogenetic distances were calculated by the NJ method using the same software.
Results and Discussion
Prophages are prevalent in S. mutans genomes
Data from 171 available whole S. mutans genomic sequences were downloaded from the NCBI website and analyzed (Table S1). The PHASTER web server was used to identify and annotate putative prophage regions within all S. mutans genomes. Thirty-five prophage-like elements were identified from 24 S. mutans genomes (13.45%) (Table 1). The genome sizes of S. mutans prophages ranged from approximately 4.7 to 68.2 kilobases, and the GC content varied between 35.62 and 44.56%. Only three prophages (phismuNLML9-1, phismuN66-1, and phismu24-1) appeared to represent complete phages with intact genomes. The remaining prophages were incomplete or questionable. The genomes of S. mutans NG8, S. mutans R221, S. mutans M230, S. mutans N29, S. mutans NLML9, S. mutans N66, and S. mutans 24 were polylysogenic. As many S. mutans genomes have prophages, and clustered regularly interspaced palindromic repeats (CRISPR)/CRISPR-associated (Cas9) can be viewed as a prokaryotic immune system that confers resistance to foreign genetic elements such as phages (Barrangou et al., 2007), we predict that CRISPR may be present in S. mutans genomes.
| Number | Prophages | Host | Accession/GI number | Size | No. CDS | Completeness | Region position | G + C percentage | References |
|---|---|---|---|---|---|---|---|---|---|
| 1 | phismuU159 | Streptococcus mutans UA159 | NC_004350.2 | 11.6 Kb | 17 | Incomplete | 1777567–1789234 | 35.62% | This study |
| 2 | phismu UA159-FR | Streptococcus mutans UA159-FR | NZ_CP007016.1 | 11.6 Kb | 12 | Incomplete | 1776275–1787942 | 35.62% | This study |
| 3 | phismu NG8-1 | Streptococcus mutans NG8 | NZ_CP013237.1 | 8.3 Kb | 6 | Incomplete | 178504–186829 | 37.77% | This study |
| 4 | phismu NG8-2 | Streptococcus mutans NG8 | NZ_CP013237.1 | 9.4 Kb | 9 | Incomplete | 590838–600315 | 37.55% | This study |
| 5 | phismu NG8-3 | Streptococcus mutans NG8 | NZ_CP013237.1 | 6.8 Kb | 7 | Incomplete | 737516–744365 | 38.31% | This study |
| 6 | phismu NG8-4 | Streptococcus mutans NG8 | NZ_CP013237.1 | 9.3 Kb | 9 | Incomplete | 1186265–1195569 | 37.39% | This study |
| 7 | phismu NG8-5 | Streptococcus mutans NG8 | NZ_CP013237.1 | 10.2 Kb | 9 | Incomplete | 1217598–1227802 | 38.30% | This study |
| 8 | phismu NG8-6 | Streptococcus mutans NG8 | NZ_CP013237.1 | 8.8 Kb | 7 | Incomplete | 1551469–1560279 | 41.32% | This study |
| 9 | phismu NG8-7 | Streptococcus mutans NG8 | NZ_CP013237.1 | 11.5 Kb | 12 | Incomplete | 1601756–1613341 | 33.74% | This study |
| 10 | phismu R221-1 | Streptococcus mutans R221 | AHRG00000000.1 | 8.6 Kb | 12 | Incomplete | 1956467–1965159 | 37.36% | This study |
| 11 | phismu R221-2 | Streptococcus mutans R221 | AHRG00000000.1 | 4.7 Kb | 12 | Incomplete | 1977201–1981928 | 38.11% | This study |
| 12 | phismu M230-1 | Streptococcus mutans M230 | AHRH00000000.1 | 25.5 Kb | 24 | Incomplete | 1785712–1811298 | 37.88% | This study |
| 13 | phismu M230-2 | Streptococcus mutans M230 | AHRH00000000.1 | 7.2 Kb | 18 | Incomplete | 1901561–1908828 | 40.24% | This study |
| 14 | phismu N29-1 | Streptococcus mutans N29 | AHRY00000000.1 | 29.7 Kb | 20 | Incomplete | 1128124–1157831 | 36.66% | This study |
| 15 | phismu N29-2 | Streptococcus mutans N29 | AHRY00000000.1 | 26.7 Kb | 7 | Incomplete | 1931418–1958213 | 37.56% | This study |
| 16 | phismu NFSM1 | Streptococcus mutans NFSM1 | AHSG00000000.1 | 10.3 Kb | 12 | Incomplete | 1966153–1976539 | 38.43% | This study |
| 17 | phismu SF14 | Streptococcus mutans SF14 | AHSQ00000000.1 | 19.5 Kb | 8 | Incomplete | 1677151–1696732 | 34.28% | This study |
| 18 | phismu U2A | Streptococcus mutans U2A | AHSU00000000.1 | 4.7 Kb | 12 | Questionable | 2094437–2099191 | 42.29% | This study |
| 19 | phismu21 | Streptococcus mutans 21 | AHSZ00000000.1 | 26.3 Kb | 21 | Incomplete | 1953048–1979373 | 36.14% | This study |
| 20 | phismu B | Streptococcus mutans B | AHTB00000000.1 | 21.2 Kb | 25 | Incomplete | 1632476–1653683 | 35.81% | This study |
| 21 | phismu SM1 | Streptococcus mutans SM1 | AHTD00000000.1 | 21 Kb | 11 | Incomplete | 1627422–1648498 | 38.20% | This study |
| 22 | phismu 3SN1 | Streptococcus mutans 3SN1 | AHRM00000000.1 | 18.1 Kb | 22 | Incomplete | 1972568–1990744 | 38.54% | This study |
| 23 | phismu11SSST2 | Streptococcus mutans 11SSST2 | AHRP00000000.1 | 6.9 Kb | 12 | Incomplete | 1947597–1954585 | 35.91% | This study |
| 24 | phismu NLML9-1 | Streptococcus mutans NLML9 | AHSJ00000000.1 | 68.2 Kb | 61 | Intact | 56412–124703 | 35.60% | This study |
| 25 | phismu NLML9-2 | Streptococcus mutans NLML9 | AHSJ00000000.1 | 5.6 Kb | 9 | Incomplete | 1953105–1958800 | 44.56% | This study |
| 26 | phismu N66-1 | Streptococcus mutans N66 | AHSM00000000.1 | 28.2 Kb | 29 | Intact | 1120823–1149060 | 35.12% | This study |
| 27 | phismu N66-2 | Streptococcus mutans N66 | AHSM00000000.1 | 6.4 Kb | 10 | Incomplete | 1907902–1914358 | 35.74% | This study |
| 28 | phismu W6 | Streptococcus mutans W6 | AHSO00000000.1 | 10.2 Kb | 11 | Incomplete | 1738562–1748818 | 35.53% | This study |
| 29 | phismu SF1 | Streptococcus mutans SF1 | AHSP00000000.1 | 20.8 Kb | 23 | Incomplete | 1543665–1564492 | 38.02% | This study |
| 30 | phismu ST6 | Streptococcus mutans ST6 | AHST00000000.1 | 16.4 Kb | 25 | Questionable | 1863533–1879945 | 37.99% | This study |
| 31 | phismu NLML1 | Streptococcus mutans NLML1 | AHSW00000000.1 | 19.9 Kb | 11 | Incomplete | 2015472–2035376 | 36.58% | This study |
| 32 | phismu 24-1 | Streptococcus mutans 24 | AHTE00000000.1 | 31.2 Kb | 38 | Intact | 1547238–1578459 | 35.98% | This study |
| 33 | Phismu 24-2 | Streptococcus mutans 24 | AHTE00000000.1 | 22.3 Kb | 14 | Questionable | 1982123–2004427 | 37.03% | This study |
| 34 | Phismu 5DC8 | Streptococcus mutans 5DC8 | AOBX00000000.1 | 11.6 Kb | 16 | Incomplete | 1741331–1753010 | 35.62% | This study |
| 35 | phismu KK21 | Streptococcus mutans KK21 | AOBY00000000.1 | 11.6 Kb | 16 | Incomplete | 1781240–1792907 | 35.62% | This study |
Sequence similarities among S. mutans prophages
Comparative genomic analyses were carried out through dot plots of 35 S. mutans prophage genomes. Dot plot analysis revealed that S. mutans prophages sorted into three clusters based on genomic similarities: Cluster A, Cluster B, and Cluster C (Fig. 1). Cluster A contains phismuU159, phismuUA159-FR, phismuKK21, phismu5DC8, and phismuN66-2. Cluster B contains phismuN66-1, phismuNLML9-1, and phismu24-1. Cluster C contains phismuNG8-3, phismuSF1, and phismu3SN1. Other S. mutans prophages, such as phismuSF14 and phismuW6, phismuM230-1 and phismuSM1, phismuN29-1 and phismuNFSM1, and phismu21 and phismuB, shared small fragments of genomic similarity, but could not be grouped into a cluster.
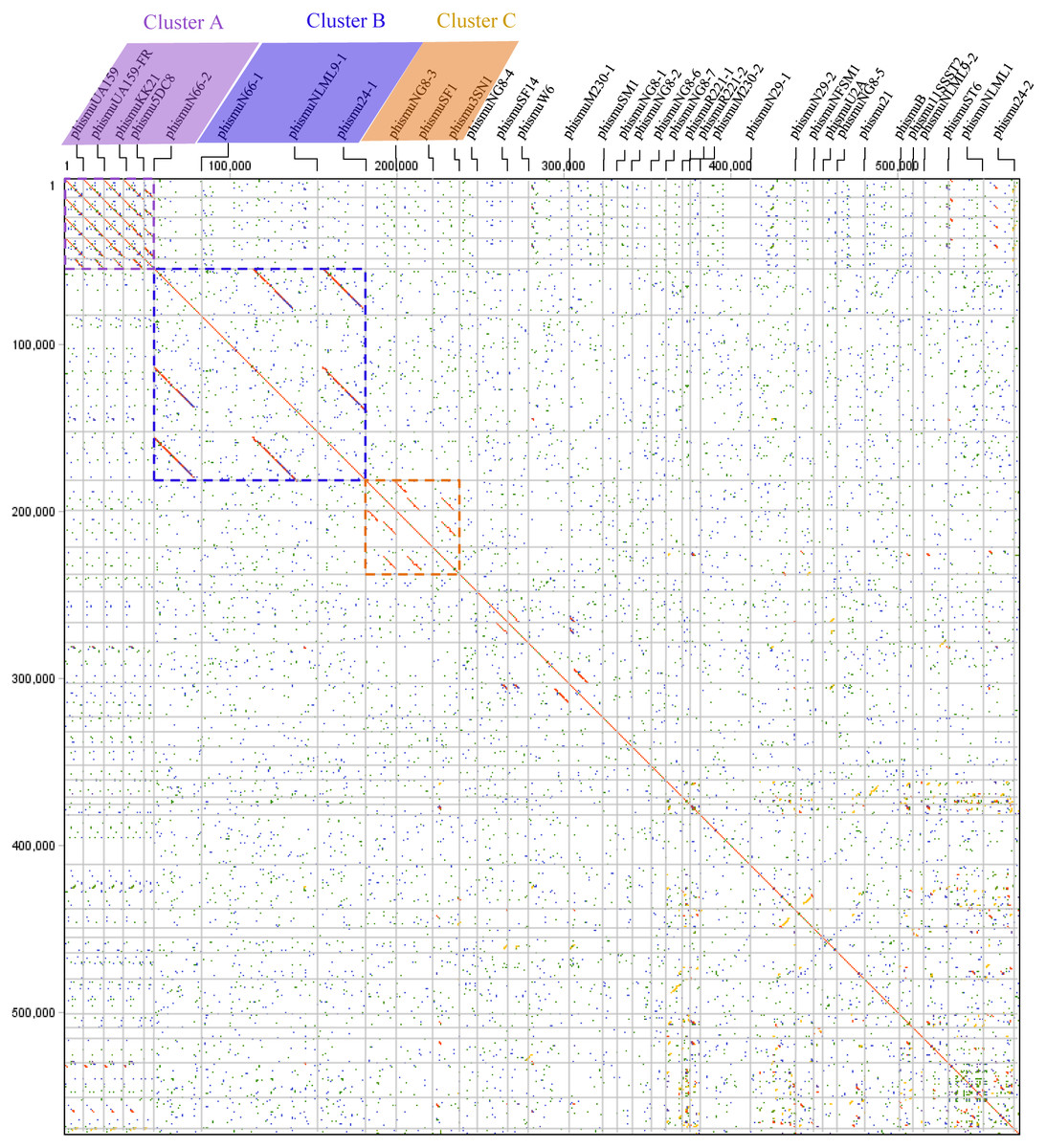
Figure 1: Dot plot matrix comparison calculated for the genomes of 35 Streptococcus mutans prophages.
Prophage genome comparison of full genomes; the main diagonal represents the alignment of a sequence with itself. Regions of local similarity or repetitive sequences give rise to further diagonal matches in addition to the central diagonal, which indicates high similarity of the prophages. The x- and y-axis indicate full genomic sequence comparisons of prophage genomes. The length of the lines represents the length of the prophage genomes. The dot plot matrix was calculated using Geneious (http://www.geneious.com, Kearse et al., 2012).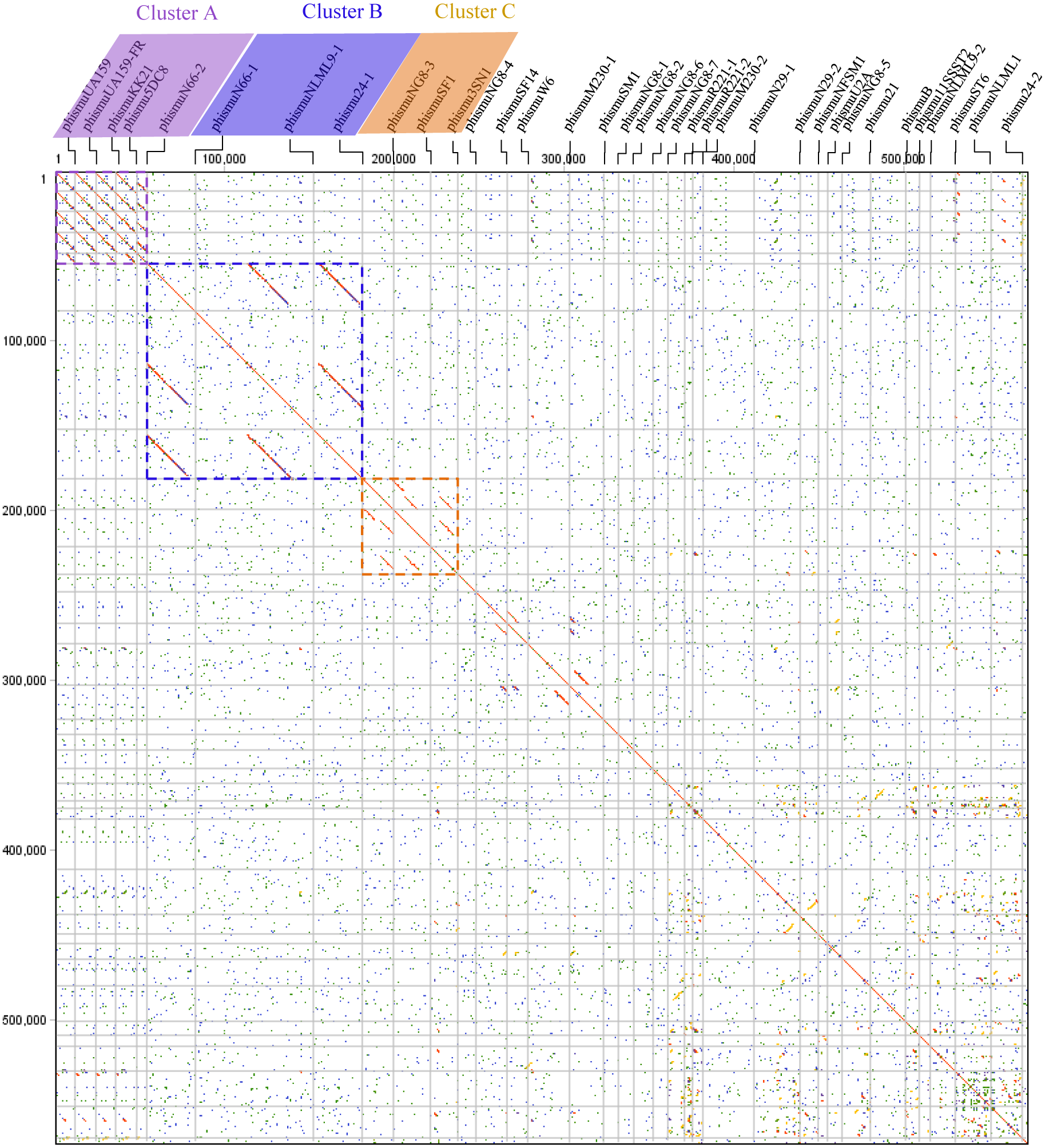{kind=link}
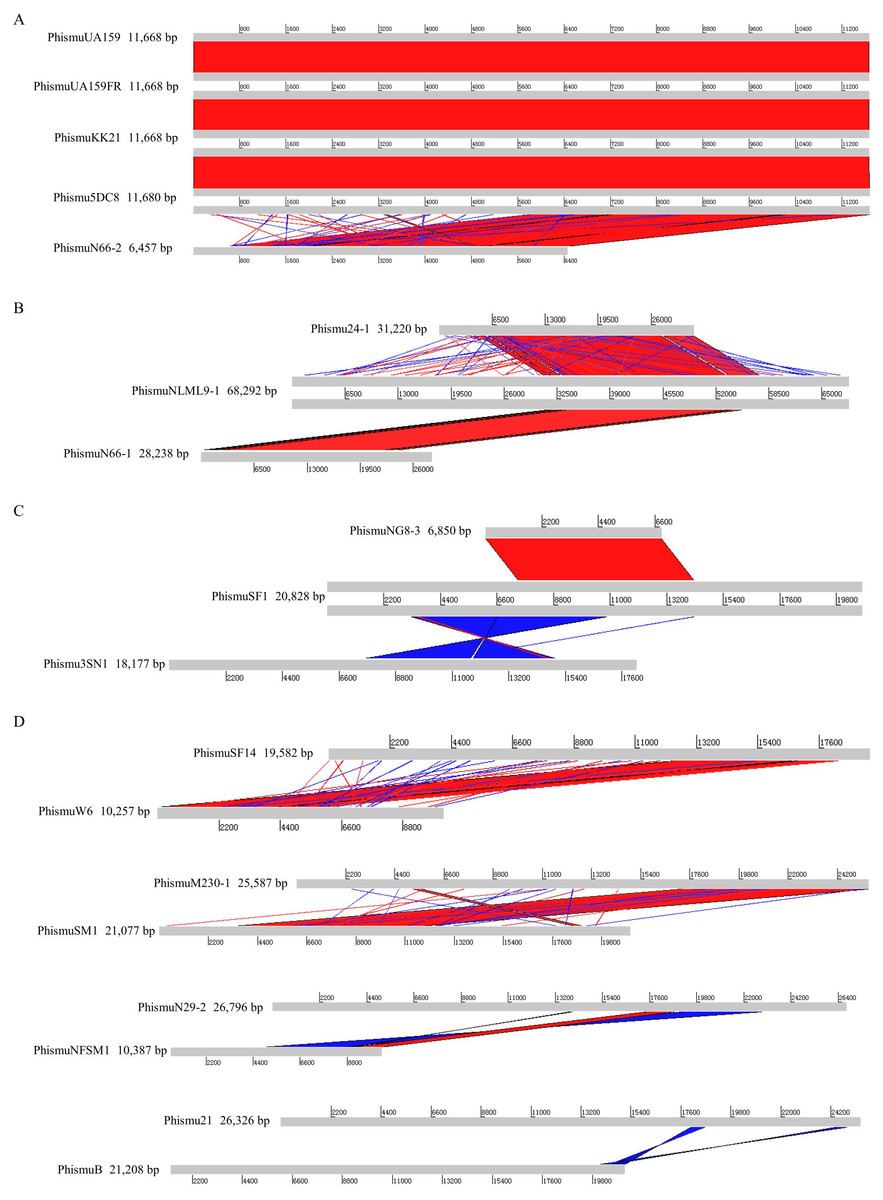
Figure 2: Genomic sequence comparison of S. mutans prophages.
Comparisons were performed using the BLASTn and Artemis Comparison Tool visualization programs. Forward and reverse matches are colored red and blue, respectively.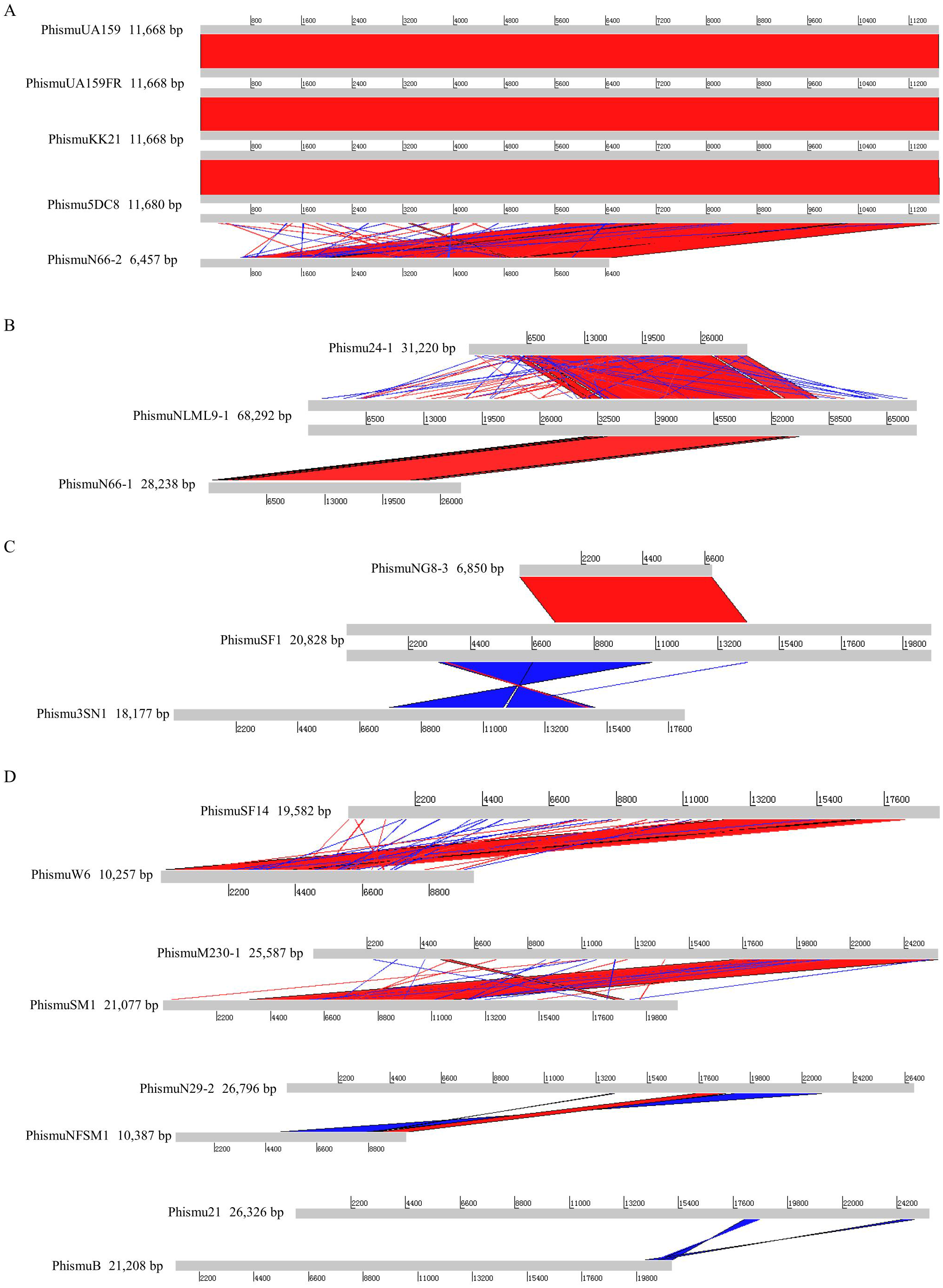{kind=link}
Comparative analysis of S. mutans prophages
Based on the similarities of their genomes, S. mutans prophages were divided into three clusters. Cluster A prophages (phismuUA159, phismuUA159-FR, phismuKK21, and phismu5DC8) shared 100% identity with one another at the genomic sequence level. Prophage phismuN66-2 shared one major region (5,854 base pairs [bp]) of sequence similarity, with 99.98% identity (Fig. 2A). Cluster B prophages (phismuN66-1, phismuNLML9-1, and phismu24-1) possessed at least 83% identity with one another, as determined by multiple genomic sequence alignments (Fig. 2B). In addition, BLASTn comparison of Cluster C (phismuSF1, phismuNG8-3, and phismu3SN1) revealed one segment (6,852 bp) with identity greater than 99% between phismuSF1 and phismuNG8-3 (Fig. 2C). Sequence comparison showed that phismuSF1 and phismu3SN1 shared two major regions of reverse complementary sequences (4,253 and 3,352 bp) with a similarity of 99.41 and 98.6% identity, respectively (Fig. 2C). Prophages belonging to individual clusters were more closely related to one another than to phages in the other clusters (Fig. S1). Other S. mutans prophages such as phismuSF14 and phismuW6 shared two major sequence (4,199 and 1,427 bp) similarities of 99.19 and 98.8% identity, respectively. PhismuM230-1 and phismuSM1 shared one major sequence (8,367 bp) similarity of 99.19% identity. PhismuN29-1 and phismuNFSM1 shared one major reverse complementary sequence (3,765 bp) similarity of 99.15% identity and one small sequence (1,291 bp) similarity of 99.15% identity. Phismu21 and phismuB shared two small reverse complementary sequence (566 and 471 bp) similarities of 99.82 and 97.66% identity, respectively (Fig. 2D).
Phylogeny of S. mutans prophages
To understand how S. mutans prophages are related to one another, a genome phylogenetic tree was constructed based on the complete genomic sequences of some S. mutans prophages, including Cluster A, Cluster B, Cluster C, and three previously reported S. mutans phages M102 (Delisle & Rostkowski, 1993), M102AD (Delisle et al., 2012), and ϕAPCM01 (Dalmasso et al., 2015). The same grouping patterns and relationships were observed among the three large clusters (Clusters A, B, and C) as in the phylogenetic tree (Fig. 3). Cluster A consisted of phismuUA159, phismuUA159-FR, phismuKK21, phismu5DC8, and phismuN66-2. PhismuUA159, phismuUA159-FR, phismuKK21, and phismu5DC8 showed the closest distinct branch in the phylogenetic tree. Cluster B consisted of M102, M102AD, ϕAPCM01, phismuN66-1, phismuNLML9-1, and phismu24-1. These results suggested that the three sequenced S. mutans phages (M102, M102AD, and ϕAPCM01) belonged to Cluster B. Cluster C consisted of phismuSF1, phismuNG8-3, and phismu3SN1.

Figure 3: Phylogenetic analysis of S. mutans (pro)phage genomic sequence alignment.
A phylogenetic tree of the genomic sequences of S. mutans (pro)phages was constructed using the neighbor-joining method. The phages were classified into three clusters, showing the phylogenetic relationships among the (pro)phage genomic sequences. Scale bar indicates 0.1 substitutions per site.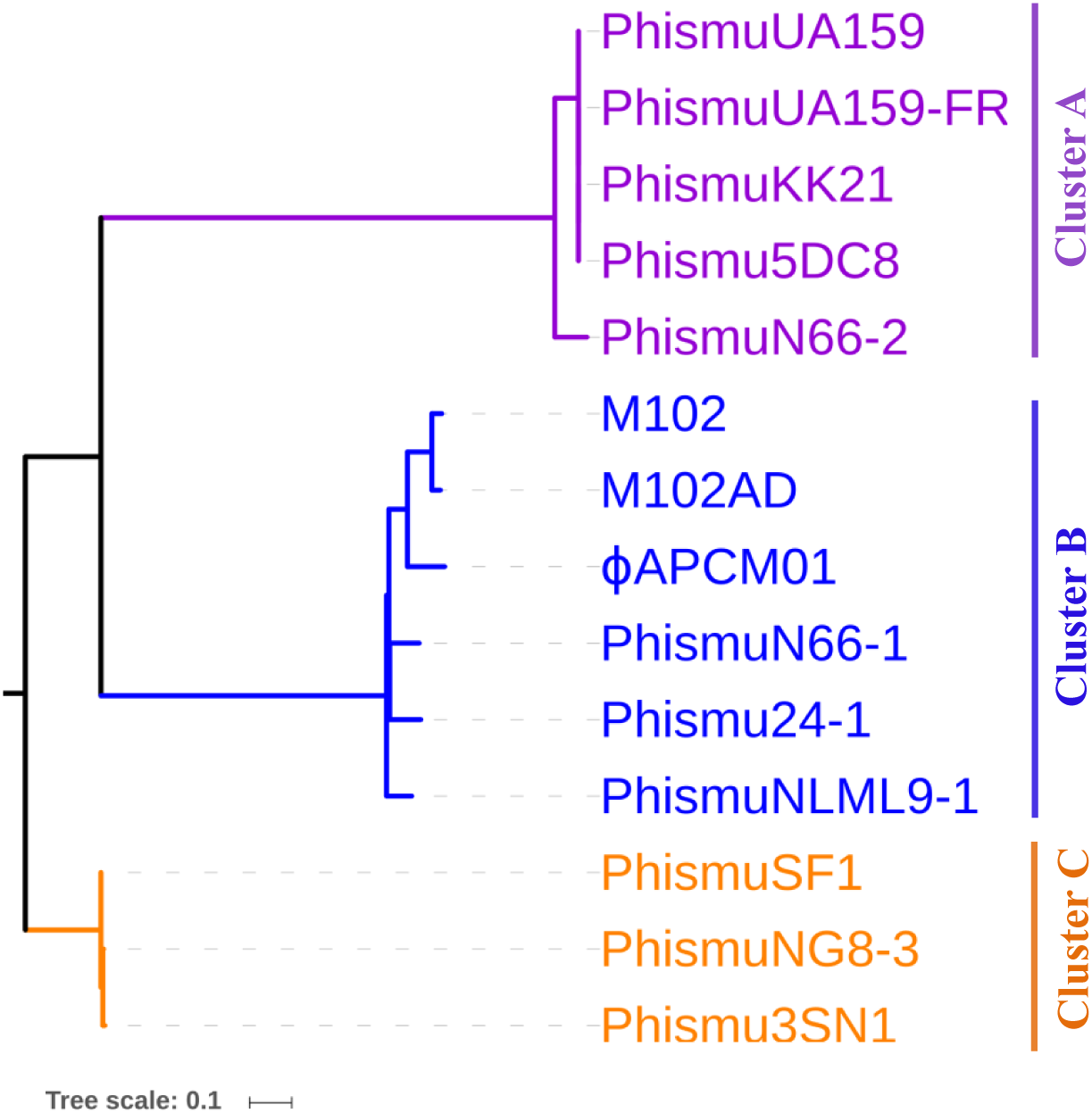{kind=link}
Comparative analysis between M102AD and S. mutans prophages
The S. mutans phage M102AD, which has a genome length of 30,664 bp and was isolated at the University of Maryland, was chosen as a reference phage, because it has been sequenced and well annotated (Delisle et al., 2012). The prophages phismuN66-1, phismuNLML9-1, and phismu24-1 all shared sequence similarity with M102AD (Fig. 4). The linear genomic comparison showed that phismu24-1 shared two major sequence (1,199 and 466 bp) similarities of 84 and 83.2% identity, respectively, with M102AD at the nucleotide level. BLASTn comparison of phismuNLML9-1 and M102AD revealed three major sequences (666, 454, and 423 bp) with 85.6, 82.9, and 82.7% identity at the nucleotide level. PhismuN66-1 shared three major sequences (749, 473, and 124 bp) with 85.6, 83.6, and 84.2% identity in comparison with M102AD genomes.

Figure 4: Sequence comparisons of M102AD with phismu24-1, phismuNLML9-1, and phismuN66-1.
Comparisons were performed using the BLASTn and Artemis Comparison Tool visualization programs. Forward and reverse matches are colored red and blue, respectively.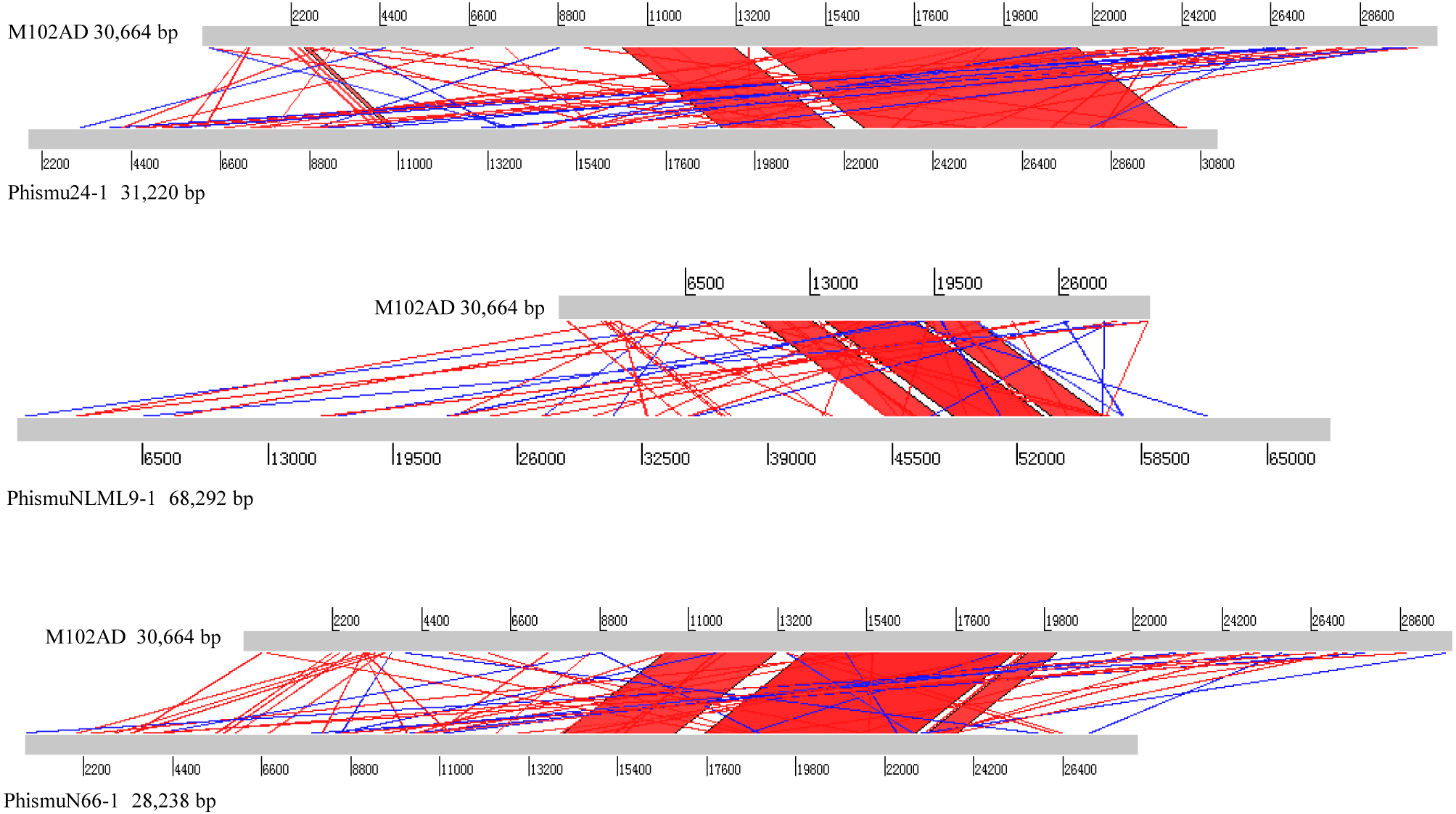{kind=link}
Summary of features of S. mutans prophage genomic sequences
Three intact prophages (phismu NLML9-1, phismu N66-1, and phismu 24-1) were identified in S. mutans, and all three prophages in Cluster B closely resembled the genome of M102AD. All of the ORFs of the prophages were predicted and annotated by PHASTER, GeneMarkS, and BLASP. PhismuNLML9-1, phismu24-1, and phismu66-1 exhibited the characteristic modular arrangement of the M102AD phage, including packaging and structural modules, integrase module, host lysis module, DNA replication/recombination module, transcriptional regulatory module, other protein module, and hypothetical protein module. The integrase gene was identified only in prophage phismuNLML9-1 (Fig. 5).
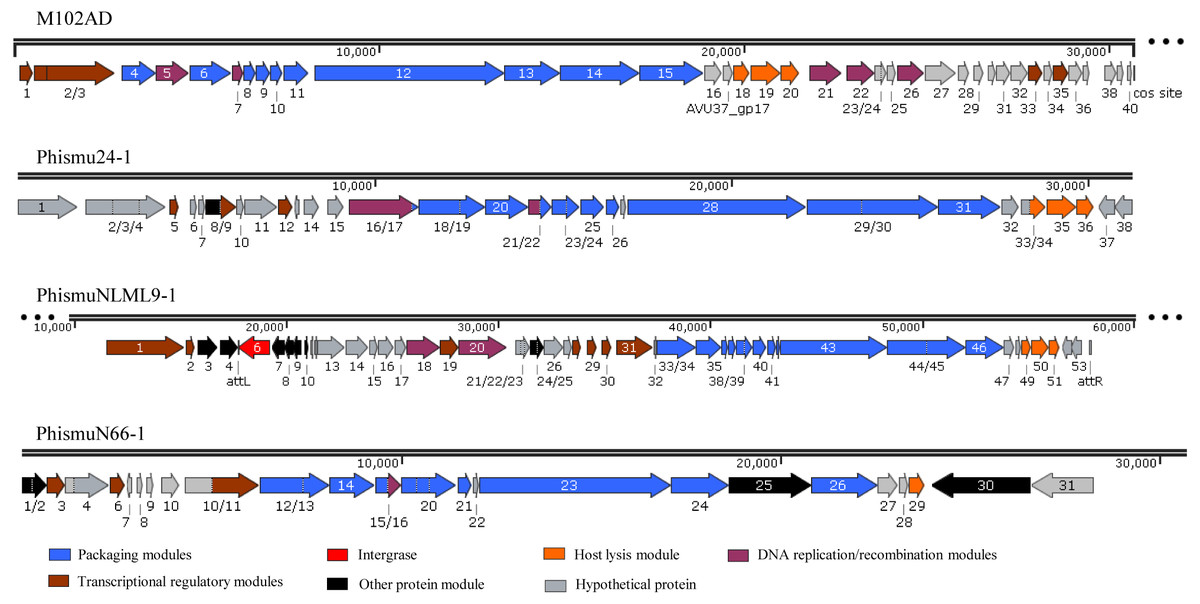
Figure 5: Genetic organization of the open reading frame (ORF) regions in the prophages M102AD, phismu24-1, phismuNLML9-1, and phismuN66-1.
Prophage genes are grouped into seven major gene cluster modules: packaging and structural modules, integrase, host lysis module, DNA replication/recombination module, transcriptional regulatory module, other protein module, and hypothetical protein module. Corresponding genes are indicated with the same color. The line numbers indicate genomic position. The predicted ORF orientations are indicated by horizontal arrows, and arrow numbers indicate the ORF region in the genomic sequence. The figure was drawn using SnapGene (http://www.snapgene.com; GSL Biotech, Chicago, IL, USA).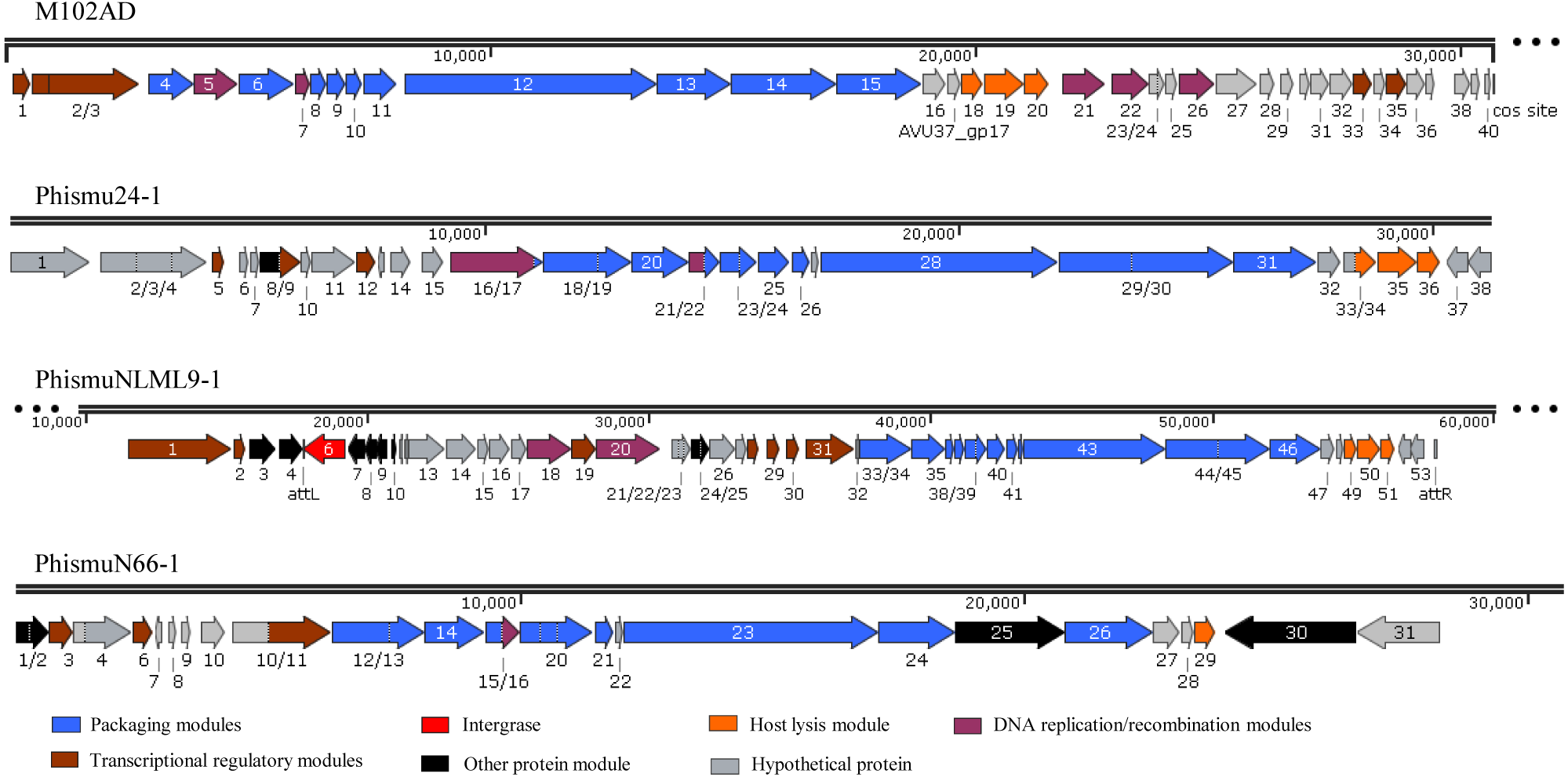{kind=link}
A total of 37 ORFs were identified in the genome of phismu24-1 (Table S2). Of the 37 ORFs, 20 were assigned a putative function. No transfer RNA (tRNA) was found in the genome of phage phismu24-1. Three significant host lysis module regions were observed in ORF 34–36. ORF 34 encodes a putative holin protein, which can form pores in cytoplasmic membranes and release toxins and other proteins or contribute to biofilm formation (Saier & Reddy, 2015). ORF 35 and ORF 36 encode putative endolysin proteins, which can digest the bacterial cell wall for phage progeny release and may have the potential to be used as antibacterial agents (Fenton et al., 2010; Fischetti, 2008). A major head and major tail protein were identified from the products of ORF 20 and ORF 25, respectively. ORF 16 and ORF 21 are predicted to be DNA replication modules, the products of which are similar to the putative large subunit of terminase and the putative DNA packaging protein.
The prophage phismuNLML9-1 contains 54 phage-related genes (Table S3). Most of the ORFs of phismuNLML9-1 are flanked by a 13-bp repeat, indicative of attL and attR sites (Fig. 5). Most temperate phages enter the lytic cycle depending on the integrase gene, which functions in chromosomal integration and excision (Smith & Thorpe, 2002). Integrase genes were identified in ORF 6 of the prophage phismuNLML9-1, suggesting that phismuNLML9-1 has the ability to enter a lysogenic replication cycle.
No putative tRNA or transfer-messenger RNA was recognized. A putative holin protein was encoded by ORF 34 and a putative endolysin by ORF 35 and ORF 36. ORF 18, ORF 9, and ORF 20 are predicted to encompass the replication module.
The phismu66-1 prophage genome contains 31 ORFs (Table S4). ORF 30 encodes the ABC transporter or permease protein, which functions as a multiple sugar metabolism transporter and is a promising target for antimicrobial strategies in S. mutans (Nagayama et al., 2014). ORF 25 encodes a host specificity protein and ORF 29 encodes a lysin-holin protein. In addition, the packaging and structural modules contained ORF 12, ORF 13, ORF 15, ORF 17–21, ORF 23, ORF 24, and ORF 26, and the transcriptional regulatory module was encoded by ORF 3, ORF 6, and ORF 11.
Horizontal gene transfer plays an important role in the adaptation and evolution of prokaryotes, and bacteriophages, as mobile genetic elements, enable horizontal gene transfer. In our study, we found that ORF 4 of phismuNLML9-1 and ORF 30 of phismu66-1 both encode an ABC transporter/permease protein, which is a virulence protein associated with the development of spontaneous resistance to compound 103 in S. aureus strains (Morisaki et al., 2016). Many unknown functional hypothetical proteins may play an important role in the acquisition of a specialized set of genes via prophages and horizontal transfer in S. mutans.
Mapping of the genomic location and comparative analysis of S. mutans prophages
We oriented and mapped the ORFs located in the genomic sequences of M102AD, phismu24-1, phismuNLML9-1, and phismuN66-1 (Fig. 6). A total of nine ORFs were located in the M102AD, phismuNLML9-1, and phismu24-1 conserved regions, and seven ORFs were located in the conserved regions of phismuN66-1. The major conserved regions of M102AD included ORFs 12–20, which encode a putative tape measure protein, putative tail protein, putative receptor-binding protein, putative minor structural protein, hypothetical protein, hypothetical protein, putative holin, putative endolysin, and putative endolysin, respectively. According to their putative functions, the ORFs were assigned to packaging/structural modules, hypothetical protein modules, and the host lysis module (Tables S5 and S6). A total of nine ORFs were identified in the conserved regions of phismu24-1, including ORF 28 and 29 (putative tail component proteins), ORF 30 (tail-host specificity protein), ORF 31 (tail protein), ORF 32 and 33 (hypothetical proteins), ORF 34 (putative holin), and ORF 35 and 36 (putative endolysins). These ORFs shared a region varying between 57.68 and 95% identity with M102AD (Table S5). The phismuNLML9-1 sequence contained nine ORFs similar to M102AD: ORF 43 and 44 (putative tail component proteins), ORF 45 (tail-host specificity protein), ORF 46 (tail protein), ORF 47 and 48 (hypothetical proteins), ORF 49 (putative holin), ORF 50 (hypothetical protein), and ORF 51 (putative endolysin). The protein sequence identity of these ORFs varied between 57.73 and 94% with M102AD (Table S5). The phismuN66-1 sequence contained seven ORFs similar to M102AD: ORF 23 and 24 (putative tail component proteins), ORF 25 (tail-host specificity protein), ORF 26 (tail protein), ORF 27 and 28 (hypothetical proteins), and ORF 29 (putative holin). The protein sequence identity of the ORFs varied between 62 and 95% with M102AD. PhismuN66-1 lost two putative endolysin ORFs compared to other prophages (Table S5).
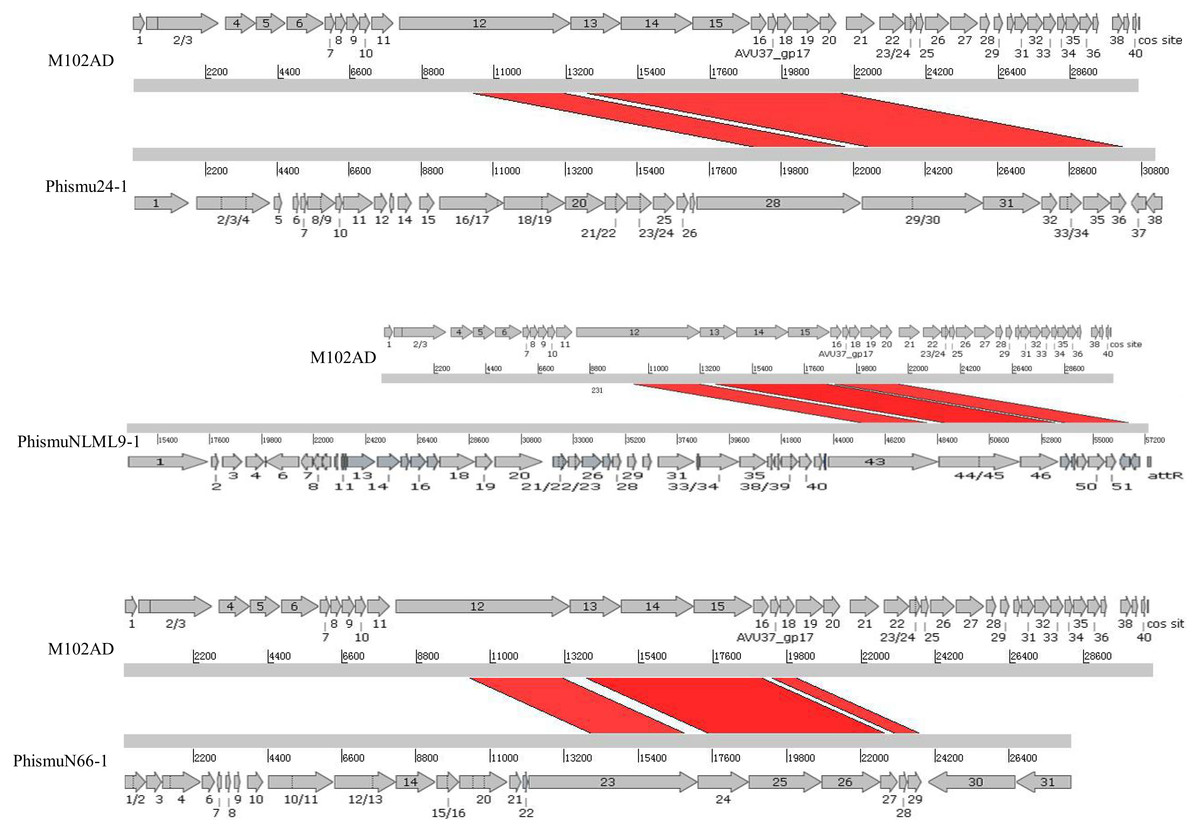
Figure 6: Schematic representation of the genomic organization and ORF regions of S. mutans phage M102AD compared to the prophages phismu24-1, phismuNLML9-1, and phismuN66-1.
The lines represent phage/prophage genomes, and arrows represent ORFs. Regions connected by red shading represent conserved genomic identity.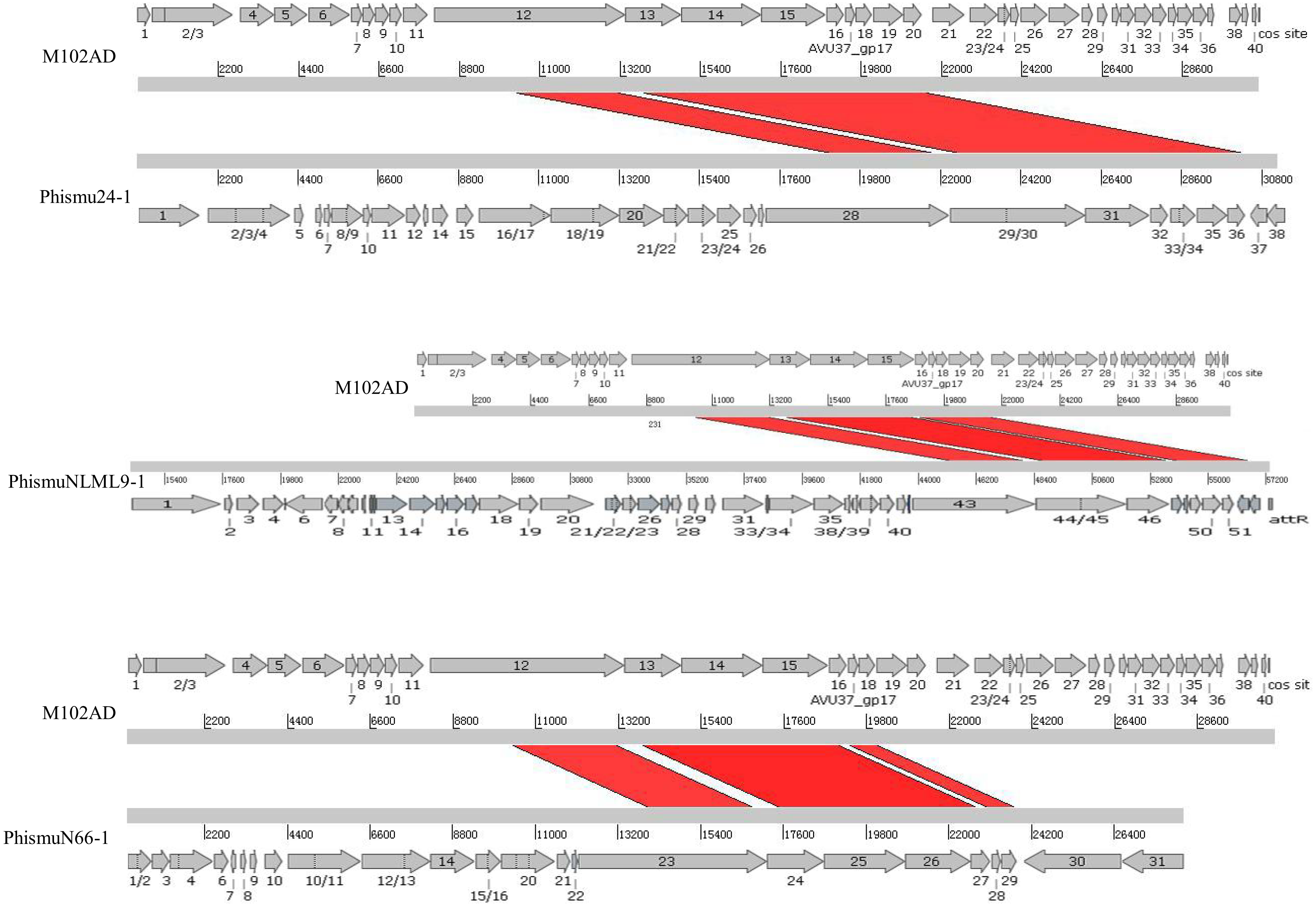{kind=link}
Conclusions
In conclusion, our genome sequencing data analyses identified 35 prophage-like elements present in the genome of S. mutans, all of which were identified for the first time. Genomic analysis of prophages revealed that those belonging to the same cluster displayed sequence similarities. The genomes and genetic information of phismuNLML9-1, phismu24-1, and phismu66-1 prophages were analyzed, identifying putative ORFs and functional regions. To the best of our knowledge, this is the first systematic analysis of S. mutans prophages.
Supplemental Information
Putative prophages detected in Streptococcus mutans strain using PHAST
Genome sequence comparison of representative clusterA, clusterB and clusterC prophages
Comparisons were performed using the Blastn and Artemis Comparison Tool visualisation programs. Forward and reverse matches are coloured in red and blue, respectively.