Electrochemical Behavior and Electrodeposition of Sn Coating from Choline Chloride–Urea Deep Eutectic Solvents

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
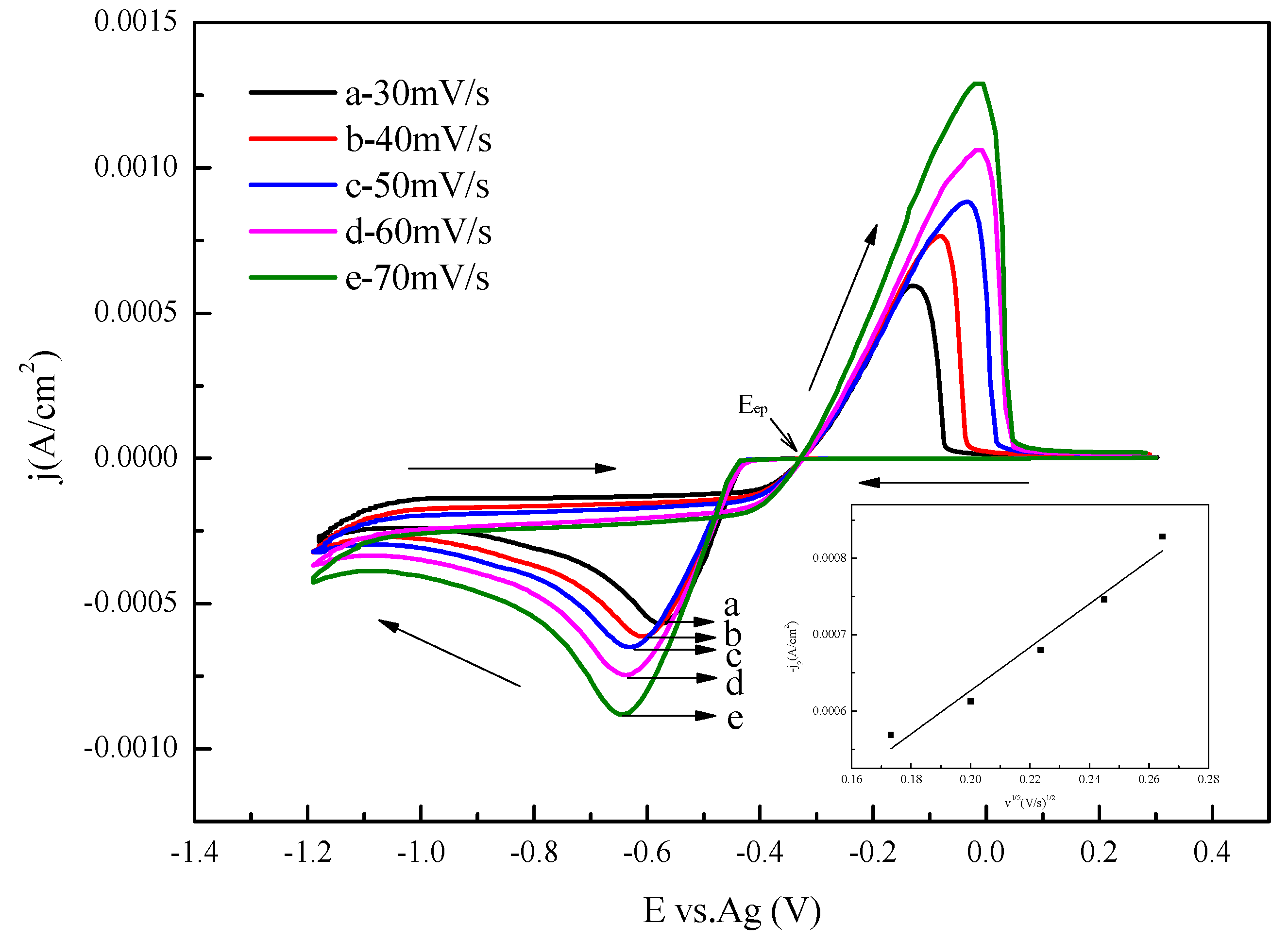
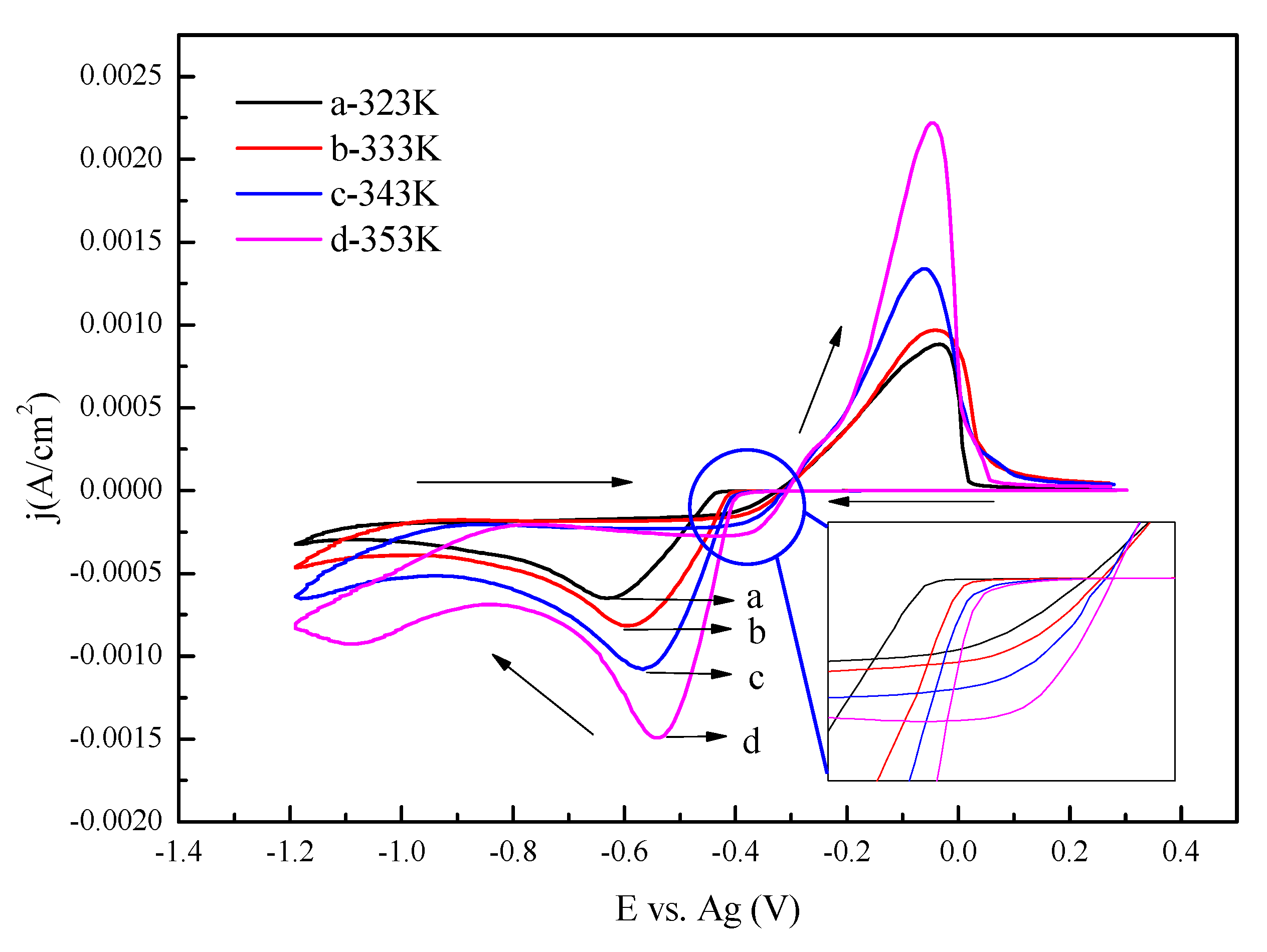
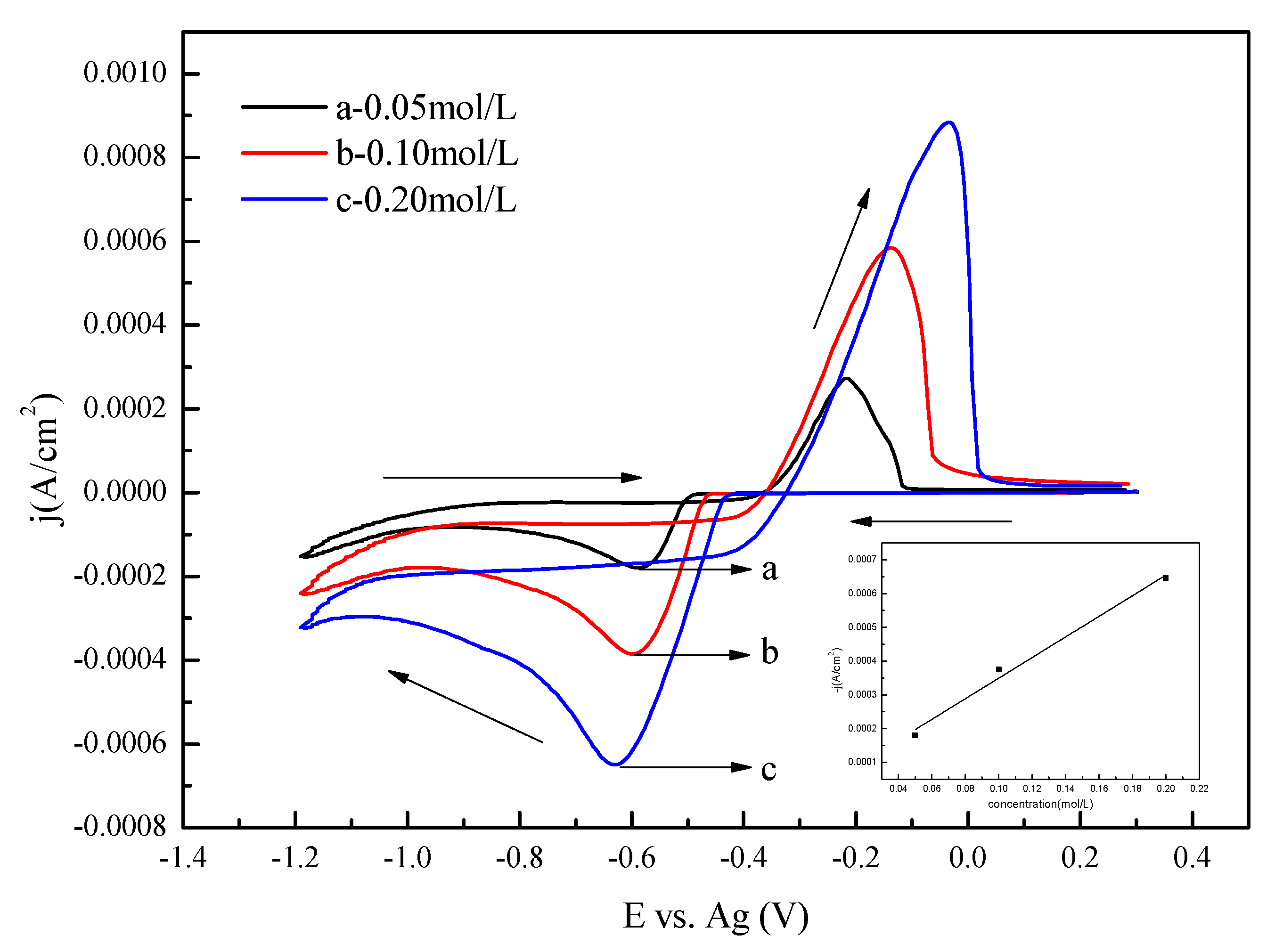
3.1. Cyclic Voltammetry
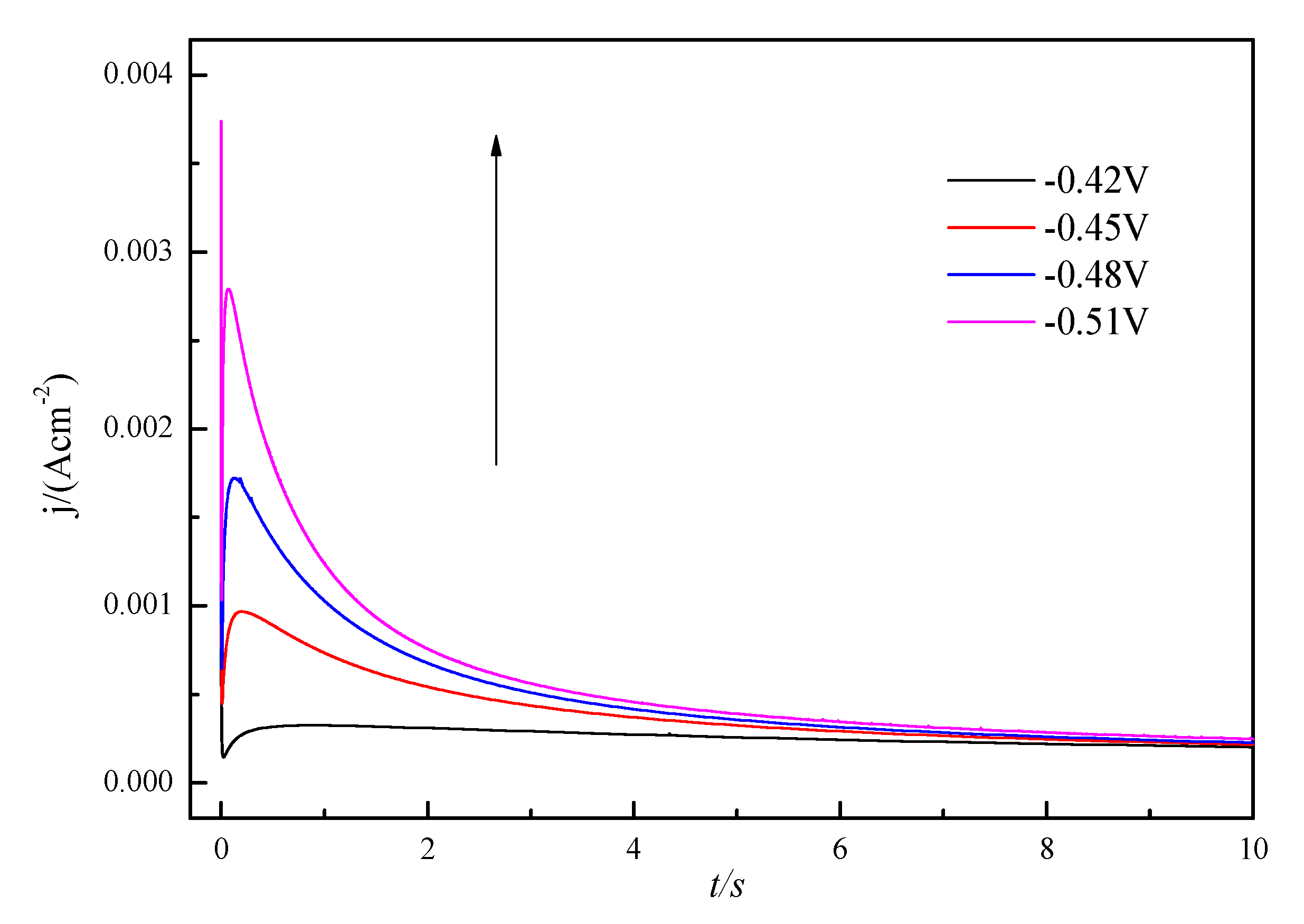
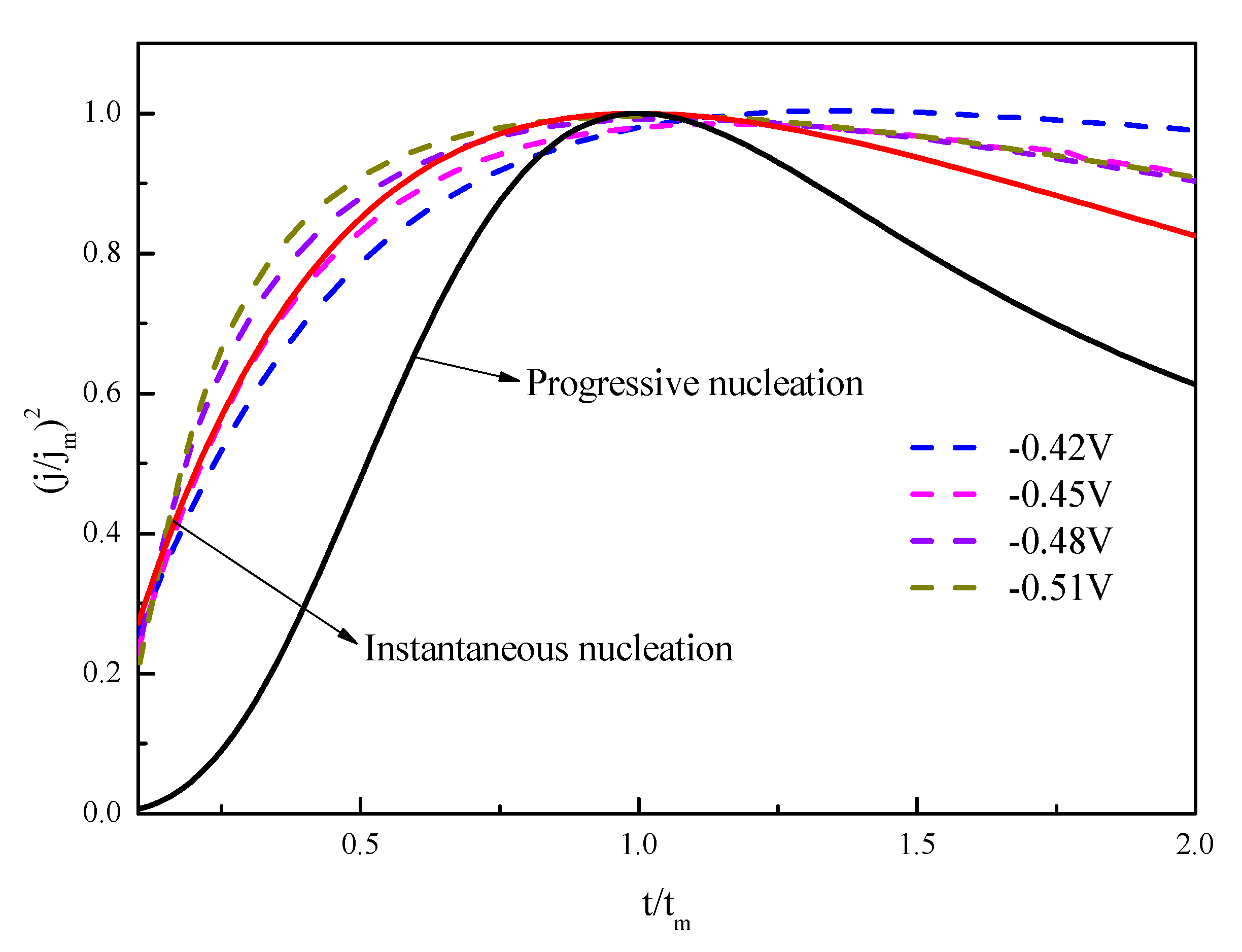
3.2. Nucleation and Growth Mechanism
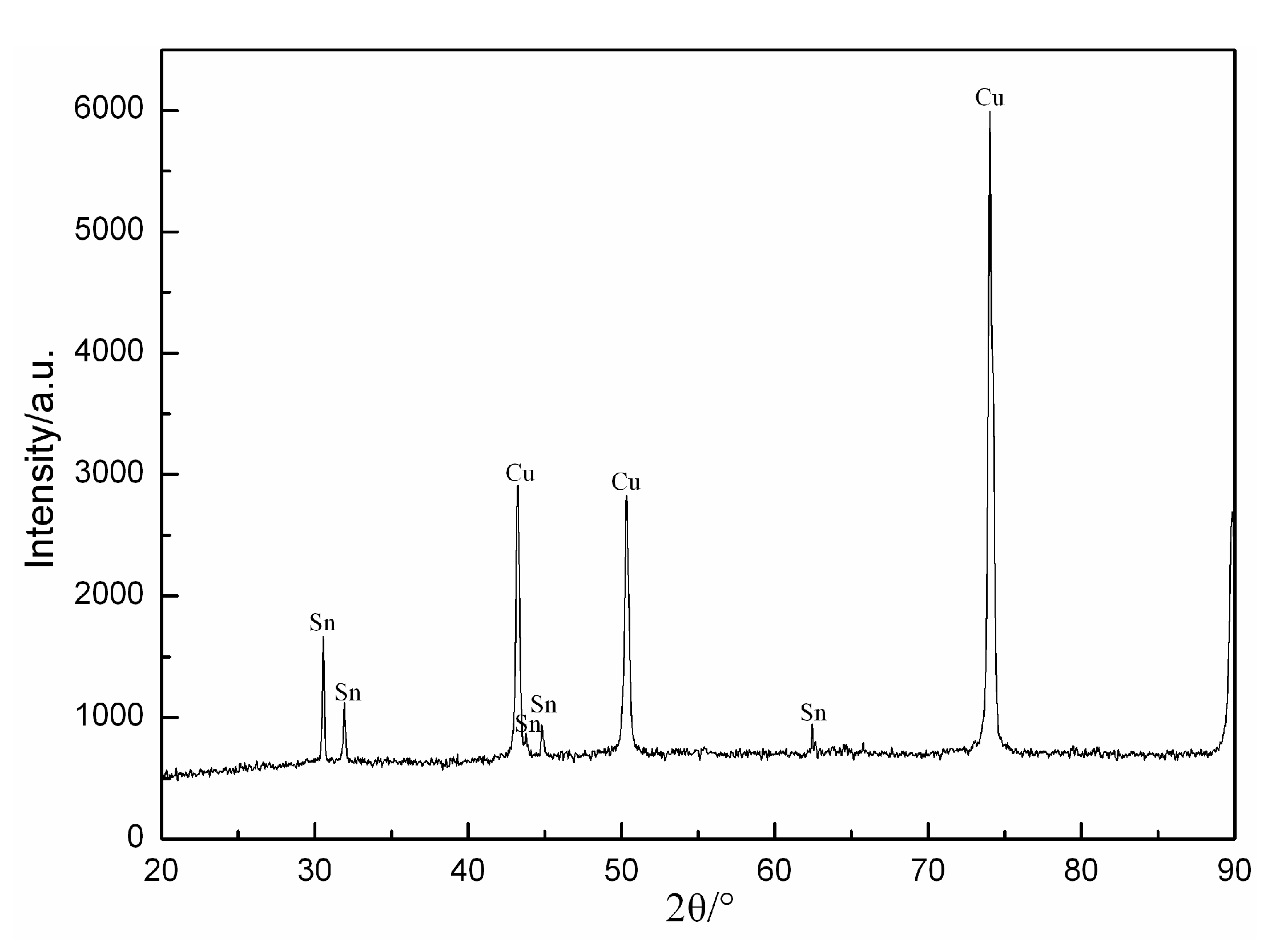
3.3. Phase Composition and Morphology of Sn Coating
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dean, R.R.; Thwaites, C.J. Tinplate and tin coating technology. JOM 1987, 39, 42–45. [Google Scholar] [CrossRef]
- Azpeitia, L.A.; Gervasi, C.S.; Bolzán, A.E. Electrochemical aspects of tin electrodeposition on copper in acid solutions. Electrochim. Acta 2019, 298, 400–412. [Google Scholar] [CrossRef]
- Walsh, F.C.; Low, C.T.J. A review of developments in the electrodeposition of tin. Surf. Coat. Technol. 2016, 288, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.; Kim, S. Study on the thermal treatment conditions for fabricating open-cell structure Cu–Sn alloy foams. J. Alloys Compd. 2021, 853, 1–10. [Google Scholar] [CrossRef]
- Gupta, A.; Srivastava, C. Nucleation and growth mechanism of tin electrodeposition on grapheme oxide: A kinetic, thermodynamic and microscopic study. J. Electroanal. Chem. 2020, 861, 1–10. [Google Scholar] [CrossRef]
- Wu, L.; Cobley, J. Investigation into the effects of magnetic agitation and pulsed current on the development of Sn–Cu alloy electrodeposits. Thin Solid Film. 2019, 683, 118–127. [Google Scholar] [CrossRef]
- Low, C.T.J.; Walsh, F.C. The stability of an acidic tin methanesulfonate electrolyte in the presence of a hydroquinone antioxidant. Electrochim. Acta 2008, 53, 5280–5286. [Google Scholar] [CrossRef]
- Alesary, H.F.; Ismail, H.K.; Shiltagh, N.M.; Alattar, R.A.; Ahmed, L.M.; Watkins, M.J.; Ryder, K.S. Effects of additives on the electrodeposition of Zn–Sn alloys from choline chloride/ethylene glycol-based deep eutectic solvent. J. Electroanal. Chem. 2020, 874, 1–11. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Mckenzie, K.J.; Ryder, K.S. Deep eutectic solvents formed between choline chloride and carboxylic acids: Versatile alternatives to ionic liquids. J. Am. Chem. Soc. 2004, 126, 9142–9147. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R. Ionic liquids based upon metal halide/substituted quaternary ammonium salt mixtures. Inorg. Chem. 2004, 43, 3447–3452. [Google Scholar] [CrossRef]
- Ismail, H.K. Electrodeposition of a mirror zinc coating from a choline chloride-ethyleneglycol-based deep etutectic solvent modified with methyl nicotinate. J. Electroanal. Chem. 2020, 876, 1–10. [Google Scholar] [CrossRef]
- Bohlen, B.; Wastl, D.; Radomski, J.; Sieber, V.; Vieira, L. Electrochemical CO2 reduction to formate on indium catalysts prepared by electrodeposition in deep eutectic solvents. Electrochim. Commun. 2020, 110, 1–5. [Google Scholar] [CrossRef]
- Landa-Castro, M.; Sebastián, P.; Giannotti, M.I.; Serrà, A.; Gómez, E. Electrodeposition of nanostructured cobalt films from a deep eutectic solvent: Influence of the substrate and deposition potential range. Electrochim. Acta 2020, 359, 1–12. [Google Scholar] [CrossRef]
- Karar, Y.; Boudinar, S.; Kadri, A.; Lepretre, J.C.; Benbranim, N.; Chaînet, E. Ammonium chloride effects on bismuth electrodeposition in a choline chloride-urea deep eutectic solvent. Electrochim. Acta 2020, 18, 1–37. [Google Scholar]
- Vieira, L.; Burt, J.; Richardson, P.W.; Schloffer, D.; Fuchs, D.; Moser, A.; Bartlett, P.N.; Reid, G.; Gollas, B. Tin, bismuth, and tin-bismuth alloy electrodeposition from chlorometalate salts in deep eutectic solvents. Chem. Open 2017, 6, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Cvetković, V.S.; Vukićević, N.M.; Jovićević, N.; Stevanović, J.S.; Jovićević, J.N. Aluminium electrodeposition under novel conditions from AlCl3-urea deep eutectic solvent at room temperature. Trans. Nonferr. Metal. Soc. 2020, 3, 823–834. [Google Scholar] [CrossRef]
- Higashino, S.; Abbott, A.P.; Miyake, M.; Hirato, T. Iron(III) chloride and acetamide eutectic for the electrodeposition of iron and iron based alloys. Electrochim. Acta 2020, 351, 1–7. [Google Scholar] [CrossRef]
- Xu, X.H.; Hussey, C.L. The electrochemistry of tin in the aluminum chloride-1-methyl-3-ethylimidazolium chloride molten salt. J. Electrochem. Soc. 1993, 140, 618–626. [Google Scholar] [CrossRef]
- Huang, J.F.; Sun, I.W. Electrochemical study of tin in zinc chloride-1-ethyl-3-methylimidazolium chloride ionic liquids. J. Electrochem. Soc. 2003, 150, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Pereira, N.M.; Pereira, C.M.; Silva, A.F. The effect of complex agents on the electrodeposition of tin from deep eutectic solvents. ECS Electrochem. Lett. 2012, 1, 5–7. [Google Scholar] [CrossRef]
- Ghosh, S.; Roy, S. Codeposition of Cu–Sn from ethaline deep eutectic solvent. Electrochim. Acta 2015, 183, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Anicai, L.; Petica, A.; Costovici, S.; Prioteasa, P.; Visan, T. Electrodepostition of Sn and NiSn alloys coatings using choline chloride based ionic liquids evaluation of corrosion behavior. Electrochim. Acta 2013, 114, 868–877. [Google Scholar] [CrossRef]
- Salomé, S.; Pereira, N.M.; Ferreira, E.S.; Pereira, C.M.; Silva, A.F. Tin electrodeposition from choline chloride based solvent: Influence of the hydrogen bond donors. J. Electroanal. Chem. 2013, 703, 80–87. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Mckenzie, K.J.; Ryder, K.S. Electrodeposition of zinc–tin alloys from deep eutectic solvents based on choline chloride. J. Electroanal. Chem. 2007, 599, 288–294. [Google Scholar] [CrossRef]
- Badea, M.L.M.; Cojocaru, A.; Anicai, L. Electrode processes in ionic liquid solvents as mixtures of choline chloride with urea, ethylene glycol or malonic acid. UPB Sci. Bull. Ser. B 2014, 76, 21–32. [Google Scholar]
- Haerens, K.; Matthijs, E.; Binnemans, K.; Bruggen, B.V. Electrochemical decomposition of choline chloride based ionic liquid analogues. Green Chem. 2009, 11, 1357–1365. [Google Scholar] [CrossRef]
- Wang, Y.S.; Yeh, H.W.; Tang, Y.H.; Kao, C.L.; Chen, P.Y. Voltammetric study and electrodeposition of zinc in hydrophobic room-temperature ionic liquid 1-butyl-1-methylpyrrolidinium Bis ((trifluoromethyl) sulfonyl)imide([BMP][TFSI]): A comparison between chloride and TFSI salts of zinc. J. ELectrochem. Soc. 2017, 164, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.Q.; Han, K.Q.; Yu, M.H.; Zhang, C.Y. The influence of sodium citrate and potassium sodium tartrate compound additives on copper electrodeposition. Int. J. Electrochem. Sci. 2017, 12, 6874–6884. [Google Scholar] [CrossRef]
- Castrillejo, Y.; Hernández, P.; Rodrigue, J.A.; Vegaa, M. Electrochemistry of scandium in the eutectic LiCl–KCl. Electrochim. Acta 2012, 71, 166–172. [Google Scholar] [CrossRef]
- Palomar-Pardavé, M.; Aldana-González, J.; Botello, L.E.; Arce-Estrad, E.M.; Ramírez-Silva, M.T.; Mostany, J.; Romero-Romo, M. Influence of temperature on the thermodynamics and kinetics of cobalt electrochemical nucleation and growth. Electrochim. Acta 2017, 241, 162–169. [Google Scholar] [CrossRef]
- Sakita, A.M.P.; Noce, R.D.; Fugivara, C.S.; Benedetti, A.V. On the cobalt and cobalt oxide electrodeposition from a glyceline deep eutectic solvent. Phys. Chem. Chem. Phys. 2016, 15, 25048–25057. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.M.; Shi, Z.N.; Reddy, R.G. Electrodeposition of Pb from PbO in urea and1-butyl-3-methylimidazolium chloride deep eutectic solutions. Electrochim. Acta 2017, 251, 176–186. [Google Scholar] [CrossRef]
- Hasan, M.M.; Hossain, M.E.; Mamun, M.A.; Ehsan, M.Q. Study of redox behavior of Cd(II) and interaction of Cd(II) with proline in the aqueous medium using cyclic voltammetry. J. Saudi Chem. Soc. 2012, 16, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Haque, F.; Rahman, M.S.; Ahmed, E.; Bakshi, P.K.; Shaikh, A.A. A cyclic voltammetric study of the redox reaction of Cu(II) in presence of ascorbic acid in different pH media. Dhaka Univ. J. Sci. 2013, 61, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Grujicic, D.; Pesic, B. Electrochemical and AFM study of cobalt nucleation mechanisms on glassy carbon from ammonium sulfate solutions. Electrochim. Acta 2004, 49, 4719–4732. [Google Scholar] [CrossRef]
- Yang, H.X.; Reddy, R.G. Electrochemical deposition of zinc from zinc oxide in 2:1 Urea/choline chloride ionic liquid. Electrochim. Acta 2014, 147, 513–519. [Google Scholar] [CrossRef]
- Scharifker, B.; Hills, G. Theoretical and experimental studies of multiple nucleation. Electrochim. Acta 1981, 28, 879–889. [Google Scholar] [CrossRef]
- He, W.C.; Liu, A.M.; Shi, Z.N.; Gao, B.L. Pb electrodeposition from PbO in The Urea/1-ethyl-2-methylinidazolium chlofide at room temperature. RSC Adv. 2017, 7, 6902–6910. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.; Xu, L.; Wang, C.; Li, S.; Wu, D.; Shi, Y.; Liu, F.; Xue, X. Electrochemical Behavior and Electrodeposition of Sn Coating from Choline Chloride–Urea Deep Eutectic Solvents. Coatings 2020, 10, 1154. https://doi.org/10.3390/coatings10121154
Cao X, Xu L, Wang C, Li S, Wu D, Shi Y, Liu F, Xue X. Electrochemical Behavior and Electrodeposition of Sn Coating from Choline Chloride–Urea Deep Eutectic Solvents. Coatings. 2020; 10(12):1154. https://doi.org/10.3390/coatings10121154
Chicago/Turabian StyleCao, Xiaozhou, Lulu Xu, Chao Wang, Siyi Li, Dong Wu, Yuanyuan Shi, Fengguo Liu, and Xiangxin Xue. 2020. "Electrochemical Behavior and Electrodeposition of Sn Coating from Choline Chloride–Urea Deep Eutectic Solvents" Coatings 10, no. 12: 1154. https://doi.org/10.3390/coatings10121154